Aproximación del laboratorio en el estudio de inmunodeficiencias
Las Inmunodeficiencias del sistema linfoide son trastornos con presentaciones clínicas heterogéneas. La mayoría de los pacientes presentan manifestaciones clínicas de disfunción inmunitaria, como infecciones recurrentes y autoinmunidad que con frecuencia causan daño orgánico irreversible en caso de diagnóstico tardío. Como consecuencia, se requiere un cribado de diagnóstico rápido y eficaz.
El sistema inmunológico humano desempeña un papel crucial en la defensa contra microorganismos infecciosos y tumores, así como en la eliminación de células y materiales viejos, dañados y extraños. Los defectos cuantitativos o funcionales en cualquiera de sus componentes pueden resultar en una mayor susceptibilidad a infecciones, neoplasias o trastornos autoinmunes, a menudo con consecuencias fatales. Estas denominadas enfermedades de inmunodeficiencia se clasifican en dos grupos según su etiopatogenia.
Las inmunodeficiencias primarias - IDP (congénitas) son causadas principalmente por defectos genéticos hereditarios que afectan negativamente el desarrollo y/o la función de uno o más componentes del sistema inmunológico, lo que resulta en una mayor tasa y gravedad de infecciones recurrentes, difíciles de tratar, o causadas por microorganismos oportunistas, que típicamente se manifiestan en la primera infancia y la niñez. Sin embargo, en algunas IDP puede haber una infección única en toda la vida del sujeto, manifestaciones autoinmunes u otros signos o síntomas más difíciles de relacionar con IDP como: úlceras o aftas gigantes de mucosas, rasgos dismórficos faciales, alteraciones cutáneas o del pelo (albinismo, vitiligo, manchas hipercrómicas, petequias, telangiectasias, alopecia), eritrodermias y dermatitis.
Las inmunodeficiencias secundarias o adquiridas (IDS) pueden desarrollarse en cualquier etapa de la vida como consecuencia de un daño causado al sistema inmunológico previamente normal, factores tales como desnutrición, infecciones (por ejemplo, VIH, VEB, CMV, sarampión y otros), enfermedades crónicas (diabetes, insuficiencia hepática y renal, enfermedades reumatológicas, metabólicas y otras), neoplasias, radiación, cirugía, trauma, tratamiento con fármacos inmunosupresores y quimioterápicos, etc.
En general, en cualquier infección que curse de manera inexplicable, con manifestaciones clínicas "raras" o acompañada de algunos de los signos anteriores, se debería descartar una IDP.
Los trastornos de IDP comprenden un grupo heterogéneo de afecciones hereditarias, extremadamente diversas en sus causas subyacentes, con más de 300 mutaciones individuales descriptas hasta ahora. Como resultado de tales mutaciones uno o más componentes de la respuesta inmune adaptativa o innata podría verse afectados. La mayoría de los pacientes con IDP (60-65%) tienen un defecto en el sistema linfoide que involucra linfocitos B (Li B) o linfocitos T (Li T) o ambos en combinación con otros tipos celulares.
Existen diferentes criterios de clasificación (Ej: ESID, IUIS, etc) pero la mayoría de las inmunodeficiencias primarias se pueden agrupar atendiendo su etiología:
- Deficiencias de Li B o de anticuerpos (70% de todas las IDP): causadas por defectos en ciertos estadios del desarrollo, diferenciación y/o maduración de los Li B (Agammaglobulinemia ligada al X, Inmunodeficiencia común variable, Deficiencia selectiva de IgA, Síndrome de Hiper-IgM, etc.)
- Deficiencias del Li T o celular (15% de todas las IDP): También llamada inmunodeficiencia combinada porque el Li T colabora en la activación del Li B para la generación de anticuerpos, por lo que en esta forma de inmunodeficiencia quedan afectadas ambas respuestas, la celular primariamente y la humoral secundariamente. Es la forma más grave de IDP. Incluyen Síndromes de inmunodeficiencia combinada severa (SCID) y otras inmunodeficiencias con predominio de Li T.
- Deficiencias de fagocitos, en su función y/o número, que constituye el 5% de todas las IDP.
- Deficiencias del complemento (10% de todas las IDP): causadas por defectos genéticos de proteínas del complemento de la vía clásica, alternativa o de lectinas).
Existen otros síndromes de inmunodeficiencia bien definidos que comprenden diversos desordenes acompañados de anormalidades neurológicas, vasculares, mucocutáneas, hematológicas o musculoesqueléticas con defectos tímicos (Síndrome de DiGeorge, Síndrome de Nezelof), defectos en la reparación del DNA (Ataxiateleangiectasia), Wiskott-Aldrich Síndrome (WAS), Síndrome de Hiper-IgE, candidiasis mucocutánea crónica, etc.
Las enfermedades de disregulación inmune representan un grupo de desórdenes con un fenotipo complejo compatible con enfermedades infecciosas, autoinmunes y linfoproliferativas. En este grupo se incluyen el Sindrome de Chediak-Higashi, Linfohistiocitosis hemofagocítica, Sindrome linfoproliferativo autoinmune (ALPS), Poliendocrinopatía autoinmune tipo 1 con candidiasis y síndrome de distrofia ectodérmica (APECED) y Poliendocrinopatía, Enteropatía, ligado a X (IPEX).
Pruebas de laboratorio en el diagnóstico y la evolución de las Inmunodeficiencias primarias
La valoración de la inmunocompetencia en un individuo con sospecha de inmunodeficiencia primaria o secundaria requiere de un análisis cuantitativo, cualitativo o estructural y de estudios funcionales que permitan detectar alteraciones en sus mecanismos de defensa. Un historial médico, un historial familiar y un examen físico precisos son fundamentales para desarrollar la mejor estrategia para la evaluación de laboratorio.
Las pruebas de laboratorio para el diagnóstico de las IP pueden dividirse en 4 grandes bloques o niveles: a) pruebas comunes de laboratorio; b) estudios de citometria de flujo (CF); c) pruebas funcionales de los linfocitos, y d) estudios genéticos y moleculares.
Aunque el diagnostico final depende muchas veces de estudios inmunológicos, genéticos y moleculares, lo cierto es que en un gran porcentaje de casos se puede hacer una aproximación diagnóstica inicial con un hemograma y unas inmunoglobulinas séricas. Ambas pruebas deben siempre de valorarse en el contexto de la edad del paciente. En caso de apreciarse una hipogammaglobulinemia, es siempre útil pedir un proteinograma para descartar pérdida proteica.
La sangre constituye la mejor ventana al sistema inmunitario y muchas IDP tienen expresión en sangre periférica, fundamentalmente citopenias. Es necesario prestar atención a la cifra de linfocitos en sangre, sobre todo en los lactantes. Una cifra de linfocitos < 2.500-3.000/μl en lactantes menores de un año puede ser un signo de inmunodeficiencia combinada grave, incluso en ausencia de otras manifestaciones clínicas. Una cifra en este rango obliga a repetir el hemograma para descartar esta grave enfermedad.
Las citopenias inmunes, afectando a una o más de las 3 series, son frecuentes en varias otras IDP. En la inmunodeficiencia variable común (IVC), las citopenias pueden presentarse varios años antes que otras manifestaciones clínicas de la enfermedad. También algunas IDP celulares, como el Síndrome de DiGeorge, y las deficiencias de adenosin deaminasa (ADA) o nucleótido fosforilasa pueden cursar con citopenias autoinmunes. En el síndrome de Wiskott-Aldrich (WAS), hay trombocitopenia con plaquetas que tienen un volumen plaquetario disminuido lo que es diagnóstico de la enfermedad en un varón. Más raramente, otras mutaciones asociadas a una ganancia de proteína dan lugar a la neutropenia crónica ligada al X.
A continuación se describen los principales estudios de laboratorio utilizados en la práctica clínica habitual, considerando los cuatro grandes grupos mencionados.
Evaluación de laboratorio para detectar deficiencia de anticuerpos o inmunidad humoral
Existe un número de defectos bien definidos en las células que participan en la respuesta inmune humoral, ciertas características como son el desarrollo de infecciones en las primeras etapas de la vida y fallas en la respuesta adecuada a estas, aún después de un tratamiento farmacológico, hacen pensar en algún tipo de deficiencia de Li B. La deficiencia de Li B puede ser relativamente moderada y no presentar síntomas mayores o puede llevar a una deficiencia severa con una completa pérdida en la síntesis y secreción de anticuerpos.
La evaluación de las inmunodeficiencias asociadas a anticuerpos incluyen las siguientes pruebas:
Cuantificación de inmunoglobulinas séricas (IgG, IgA, IgM)
El dosaje de IgA, IgM e IgG séricas se realiza rutinariamente en muchos laboratorios. Actualmente existen varias técnicas siendo la nefelometría la más recomendada. La evidencia avala la medición de inmunoglobulinas en el suero como el examen más adecuado al momento de sospechar déficit de anticuerpos, siendo de utilidad diagnóstica en patologías como agammaglobulinemia ligada al X (De Brutton), inmunodeficiencia común variable, déficit de IgA, entre otras.
Adicionalmente, medir los niveles de IgE total, es de utilidad ya que existen inmunodeficiencias asociadas a elevación de esta inmunoglobulina, los llamados síndromes Hiper-IgE asociados a infecciones bacterianas recurrentes y dermatitis.
Medición de anticuerpos post-vacunales
Por otra parte, unas cifras de Inmunoglobulinas normales no descartan la existencia de una deficiencia de anticuerpos. Si la clínica es sugerente, es conveniente determinar los títulos de anticuerpos naturales, como las isohemaglutininas, o inducidos, como son los anticuerpos antitetánicos. En niños mayores de 3 años, se debe valorar también la formación de anticuerpos antipolisacáridos. En muchos casos, es necesario revacunar y medir los títulos de anticuerpos pre y posvacunación.
Estas pruebas miden qué tan bien responde el sistema inmunológico a las vacunas comunes incluidas las que tienen antígenos proteicos, T-indepedendientes, (como el toxoide tetánico, toxoide diftérico) y las que tienen antígenos de carbohidratos, T-dependientes (bacterias encapsuladas, como las del pneumococco). En algunos casos, es posible que el paciente ya haya sido inmunizado con estas vacunas como parte de su cuidado normal y ya tendrá anticuerpos circulantes, mientras que en otros casos el paciente puede tener poco o ningún anticuerpo específico antes de la inmunización. El uso de diferentes tipos de vacunas es necesario porque ciertos pacientes con infecciones recurrentes (y niveles de inmunoglobulinas normales o casi normales) han sido identificados con una anomalía en la respuesta a los antígenos de carbohidratos pero una respuesta normal a los antígenos de proteínas (durante la maduración del sistema inmunológico, la respuesta a las vacunas de antígenos de carbohidratos va por detrás de la respuesta a las vacunas de antígenos proteicos).
Medición de subclases de inmunoglobulinas
Las inmunoglobulinas se han agrupado en diferentes subclases de acuerdo a diferencias que se encuentran en las cadenas pesadas. La IgG se subdivide en IgG1, IgG2, IgG3 e IgG4. Una deficiencia en las subclases de IgG puede resultar en la producción alterada de ciertos tipos de anticuerpos. La deficiencia selectiva de anticuerpos más frecuentemente identificada, es una respuesta defectuosa a antígenos de polisacáridos, como los presentes en la cápsula de neumococo, meningococo y Hemophilus influenzae tipo B, donde los niveles disminuidos de IgG2 provocan un pobre respuesta a infecciones por bacterias encapsuladas.
Isohemaglutininas
Dependiendo de la edad del paciente y su grupo sanguíneo es posible medir la presencia de anticuerpos naturales o isohemaglutininas anti-A y anti-B, predominantemente de clase IgM dirigidos contra antígenos del grupo sanguíneo. Es una prueba útil en casos de inmunodeficiencias con falla selectiva para la formación de IgM, por ejemplo el síndrome de Wiskott Aldrich.
Recuento de Li B
La citometria de flujo (se describe en mayor detalle más adelante) es también primordial en el estudio de las IDP humorales, ya que permite, de forma muy sencilla, hacer una primera aproximación diagnostica al saber si el paciente tiene o no Li B.
Evaluación de laboratorio para detectar deficiencias de la inmunidad celular y combinada
El número de poblaciones de linfocitos puede determinarse mediante citometría de flujo (CF), mientras que su función puede examinarse in vitro mediante la prueba de proliferación de linfocitos (prueba de transformación blástica) e in vivo mediante pruebas cutáneas de hipersensibilidad de tipo retardado.
Recuento de poblaciones linfocitarias
La CF es la técnica mas útil en el estudio de la mayoría de las IP, ya que permite evaluar las poblaciones y subpoblaciones celulares, así como identificar moléculas de la membrana y del citosol, e incluso valorar la función anormal de una proteína. Como escrutinio de las IP, es útil realizar un inmunofenotipo de las poblaciones celulares de la sangre mediante la identificación de marcadores celulares de superficie mediante CF. Con ello se logra la presunción diagnostica de la mayoría de IP de Li T y Li B, que posteriormente se confirma, mediante otros estudios. Es posible medir mediante citometria subpoblaciones B (pre swicht, post swicht, transicionales, etc) y T (memoria, vírgenes, activadas), subpoblaciones T helper: Th1, Th2 y TH17 para confirmar patologías en particular.
En algunas situaciones, el status T/B/NK (CD3/CD19/CD56) es suficiente para anticipar una IDP específica y provee evidencia diagnóstica de ID celular, humoral o combinada, que posteriormente se confirma mediante otros estudios. En contraste, en pacientes con reducido número absoluto de Li T o importante aumento de células NK, la causa primaria es generalmente un poco más difícil de ser dilucidada. Sin embargo, en muchas IDP, el número absoluto de células T, B y/o NK observadas son normales.
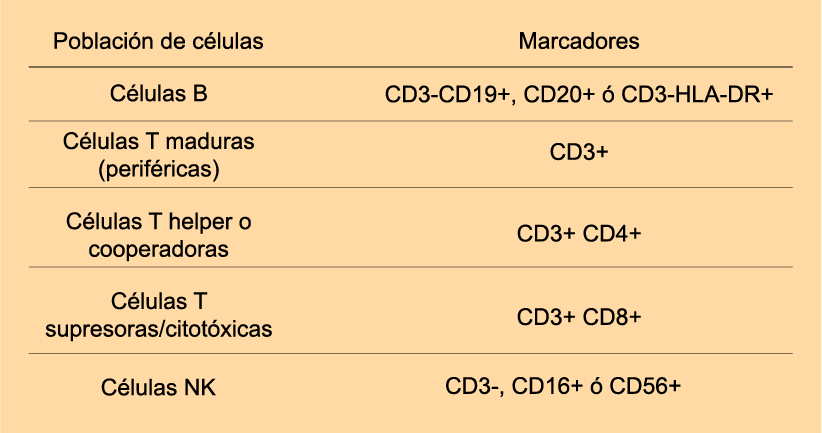
Los recuentos de Li B extremadamente bajos o ausentes son típicos de la agammaglobulinaemia ligada al X, sin embargo, la disminución o ausencia de este tipo celular también se puede encontrar en algunas inmunodeficiencias combinadas graves (disgénesis reticular, deficiencia de adenosina deaminasa, deficiencia de RAG1/2) y ocasionalmente en inmunodeficiencia variable común. El número de Li T bajos o ausentes es característico de las inmunodeficiencias combinadas, los síndromes de Di George y Wiskott-Aldrich, la ataxia-teleangiectasia y algunos otros de los síndromes de IDs bien definidos. Los recuentos bajos de Li T CD4 (helper) y una disminución de la relación de Li TCD4+/CD8+ se encuentran típicamente en inmunodeficiencia variable común y deficiencia de clase II; sin embargo, también se asocian con afecciones como la infección por VIH, algunas infecciones bacterianas y parasitarias, trauma físico, tratamiento con corticosteroides o estrés (IDS). El aumento del número de Li T citotóxicos y una mayor relación CD4+/CD8+ son típicos de una deficiencia rara de CD8, deficiencia de clase I del MHC y numerosos trastornos autoinmunes.
Una mayor presencia de células CD4-CD8- doble negativa en la sangre periférica se encuentra típicamente en síndromes autoinmunes linfoproliferativos (ALPS) causados por defectos de mediadores de la apoptosis, mientras que bajos niveles de células NK se encuentran en ciertas deficiencias combinadas (deficiencia de IL-2RG, deficiencia de JAK3) y/o deficiencias raras de células NK.
La evaluación de la función de los Li T se puede medir, provocando «in vitro» la activacion y la proliferación de los linfocitos mediante diferentes moléculas mitogenos y antigenos, que simulan la activación natural del linfocito TH.
La función de las células NK se puede evaluar mediante la detección de perforina y granzima por CF, de utilidad en la Linfohistiocitosis hemofagocitica familiar por déficit de perforina (20-30 % de los casos)
Hipersensibilidad cutánea retardada
Se considerada como una correlación in vivo de la inmunidad celular y puede ser usada como método de monitoreo para evaluar defectos en la inmunidad mediada por células. La mayoría de loscindividuos inmunocompetentes presentan una prueba positiva, mientras que los individuos con algún tipo de inmunodeficiencia presentan resultados negativos.
Evaluación de laboratorio para detectar deficiencias de los fagocitos
Una revisión cuidadosa del frotis de sangre es importante para descartar ciertas enfermedades que están asociadas con anomalías en la estructura del neutrófilo. Si el hemograma exhibe recuento normal o aumentado de neutrófilos, las pruebas siguientes se centrarían en dos posibles trastornos inmunitarios primarios: la enfermedad granulomatosa crónica (EGC) y la deficiencia de adhesión de leucocitos (LAD).
Para diagnosticar la EGC se realiza el test de DHR mediante CF que permite evaluar si está conservada la capacidad de los neutrófilos para producir el estallido respiratorio u oxidativo. Las células de los sujetos con la enfermedad no presentan fluorescencia o está muy disminuida, a diferencia de los sujetos normales. En las madres portadoras hay 2 grupos de poblaciones celulares.
El diagnostico de LAD tipo 1 implican determinar la presencia de B2-integrinas en la superficie de los neutrófilos (y otros leucocitos) por CF. Cuando esta proteína está ausente o disminuida significativamente (por mutaciones en el gen que codifica la cadena b-integrina), el movimiento de los neutrófilos hacia los sitios de infección se ve obstaculizado y produce un gran aumento en el número de estas células en la circulación, así como una mayor susceptibilidad a infecciones bacterianas cutáneas y orales.
Evaluación de laboratorio para detectar deficiencias del complemento
Evaluar los componentes del complemento y su función, aspira a determinar la concentración de los componentes individuales del complemento (C3, C4, C1q, inhibidor de C1 esterasa), y su capacidad funcional. (CH50).
Las deficiencias primarias son muy raras y en general afectan a los componentes del complemento C3 y C4, que se manifiestan en el paciente con infecciones bacterianas recurrentes. La deficiencia de inhibidor de C1 esterasa es la causa del angioedema hereditario, un trastorno autosómico dominante, que se caracteriza por el edema de los tejidos subcutáneos.

En la gran mayoría de los casos, además de estos enfoques diagnósticos descriptos, el diagnóstico molecular de las de mutaciones de IDP es una parte integral de la estrategia diagnóstica y una herramienta importante para la identificación de mutaciones específicas que causan enfermedades. Además, forman una base para un tratamiento adecuado, estimación del pronóstico, desarrollo de estrategias preventivas, asesoramiento genético y diagnóstico prenatal. La detección de defectos genéticos puede realizarse a nivel de ADN o ARN mediante varios métodos de PCR y secuenciación de ADN.
Autora: Dra. Romina P. Ranocchia - Area Inmunología, autoinmunidad y serología. Citometría de Flujo - Fares Taie Instituto de Análisis
Contacto: ranocchia@farestaie.com.ar
BIBLIOGRAFIA
- Pruebas de laboratorio en el diagnóstico de las inmunodeficiencias primarias. An Pediatr Contin. 2013;11(5):282-90
- Clinical and laboratory assessment of immunity. J ALLERGY CLIN IMMUNOL VOLUME 111, NUMBER 2
- The 2017 IUIS Phenotypic Classification for Primary Immunodeficiencies. J Clin Immunol. 2018 Jan;38(1):129-143.
- Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J Clin Immunol. 2020 Jan;40(1):66-81.
- Diagnóstico precoz de las inmunodeficiencias. Signos guía y pruebas complementarias orientativas para el pediatra. An Pediatr Contin. 2011;9(3):145-52
- https://www.farestaie.com.ar/novedades/profesionales/1288-valoracion-de-poblaciones-linfocitarias-por-citometria-de